Iso-Seq Collapse
After transcript sequences are mapped to a reference genome, isoseq collapse can be used to collapse redundant transcripts (based on exonic structures) into unique isoforms. Output consists of unique isoforms in GFF format and secondary files containing information about the number of reads supporting each unique isoform.
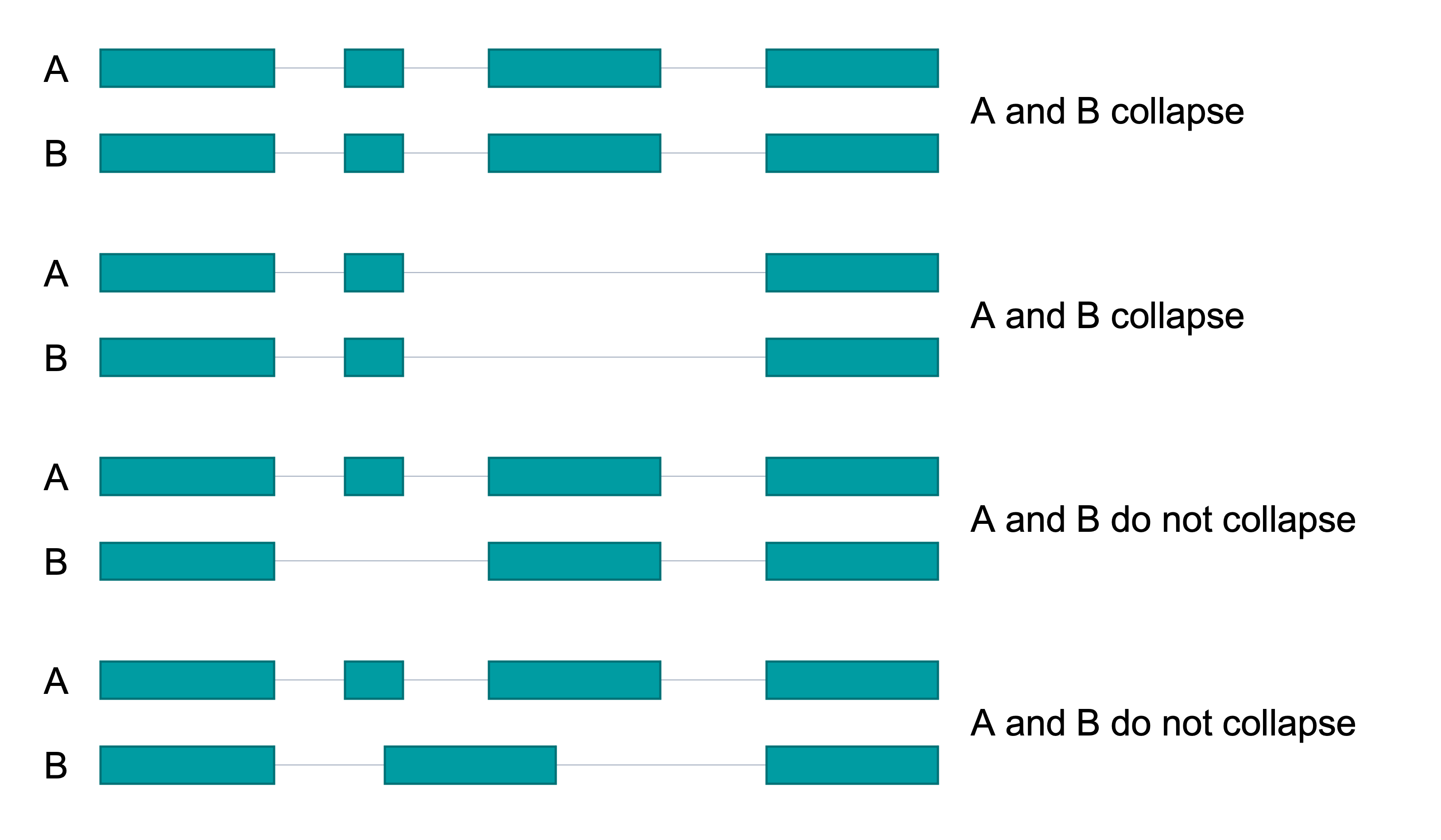
Collapse Examples
Execution
Map reads using pbmm2 before collapsing
pbmm2 align --preset ISOSEQ --sort <input.bam> <ref.fa> <mapped.bam>
Collapse mapped reads into unique isoforms using isoseq collapse.
For single-cell Iso-Seq:
isoseq collapse <mapped.bam> <collapse.gff>
For bulk Iso-Seq:
isoseq collapse --do-not-collapse-extra-5exons <mapped.bam> <flnc.bam> <collapsed.gff>
Notes:
- The optional
<flnc.bam>input is required to get the correct FLNC counts for bulk Iso-Seq in theflnc_count.txtsupplemental file. collapseby default will collapse isoforms containing 5p degradation as of version3.8.0. To turn this off--do-not-collapse-extra-5exonsshould be used. This option is recommended for bulk Iso-Seq.
Output
Collapsed transcripts are assigned unique IDs in the format PB.X.Y, where X is based solely on genomic mapping region - not exonic structure. This means X can cover multiple genes. Y represents the Y-th transcript in the X region.
Output files:
collapse.gffcontains the collapsed isoforms in gff format.*.abundance.txtcontains information about the number of FLNC reads supporting each isoform and cell barcodes if applicable. Each unique isoform has the ID format PB.X.Y, whilecount_fldenotes the number of unique molecules (after UMI deduplication) supporting the isoform, andfl_assocdenotes the number of reads (before UMI deduplication) supporting it.cell_barcodesshows the list of single cell barcodes from which the reads came from, if applicable. This file should be used for downstreampigeonsteps for Single-cell Iso-Seq.pbid count_fl fl_assoc cell_barcodes PB.1.1 2 2 ATCCATTCACCTCTGT,ATCGGCGCAGAGATGC PB.2.1 1 1 CGGACACCATTGCCGG PB.3.1 1 1 ACTTCGCGTCTAACTG*.flnc_count.txtcontains information about the number of FLNC reads supporting each isoform before any clustering or deduplication. Each unique isoform has the ID format PB.X.Y and the FLNC counts will be separated by sample if multiple samples are present. This file should be used for downstreampigeonsteps for Bulk Iso-Seq.id BioSample1 BioSample2 PB.1.1 2 2 PB.2.1 1 2 PB.3.1 1 1*.group.txtshows the grouping of redundant isoforms (based on mapped exonic structures), where the read namesmolecule/<number>denote a unique molecule after UMI deduplication and the read namestranscript/<number>denote a clustered transcript.PB.1.1 molecule/7343975,molecule/7738347 PB.2.1 molecule/14601188 PB.3.1 molecule/3998518*.read_stat.txtshows the assignment of each read (before UMI deduplication or clustering) to the final, unique isoforms PB.X.Y. Read names with the format<movie>/<zmw>/ccsindicate a CCS read, whereas<movie>/<zmw>/ccs/<start>_<end>further denotes a segment of a CCS read (S-read), likely as a result of segmentation (using, for example, Skera) of concatenated single cell libraries.id pbid m64012_220421_000242/120719489/ccs/10460_11196 PB.1.1 m64012_220421_000242/17565024/ccs/13918_14203 PB.1.1 m64012_220421_000242/161089449/ccs/1955_2888 PB.2.1 m64012_220421_000242/158664505/ccs/2488_2901 PB.3.1
Collapse FAQ
As of isoseq3 v3.8.0 collapse has algorithmic updates. These updates include performance improvements and updates to isoform collapse logic.
What is new in v3.8.0 and later?
Collapsing extra 5p exons
For applications like single-cell Iso-Seq where there is a higher percentage of 5p truncated isoforms, it is useful to collapse isoforms that have a matching exon structure with the exception of extra 5p exons. Previous versions of collapse did not merge isoforms with extra 5p exons. As of v3.8.0, collapse will merge these isoforms by default. To not allow merging isoforms with extra 5p exons, use --do-not-collapse-extra-5exons. This option is used in the bulk Iso-Seq workflow.
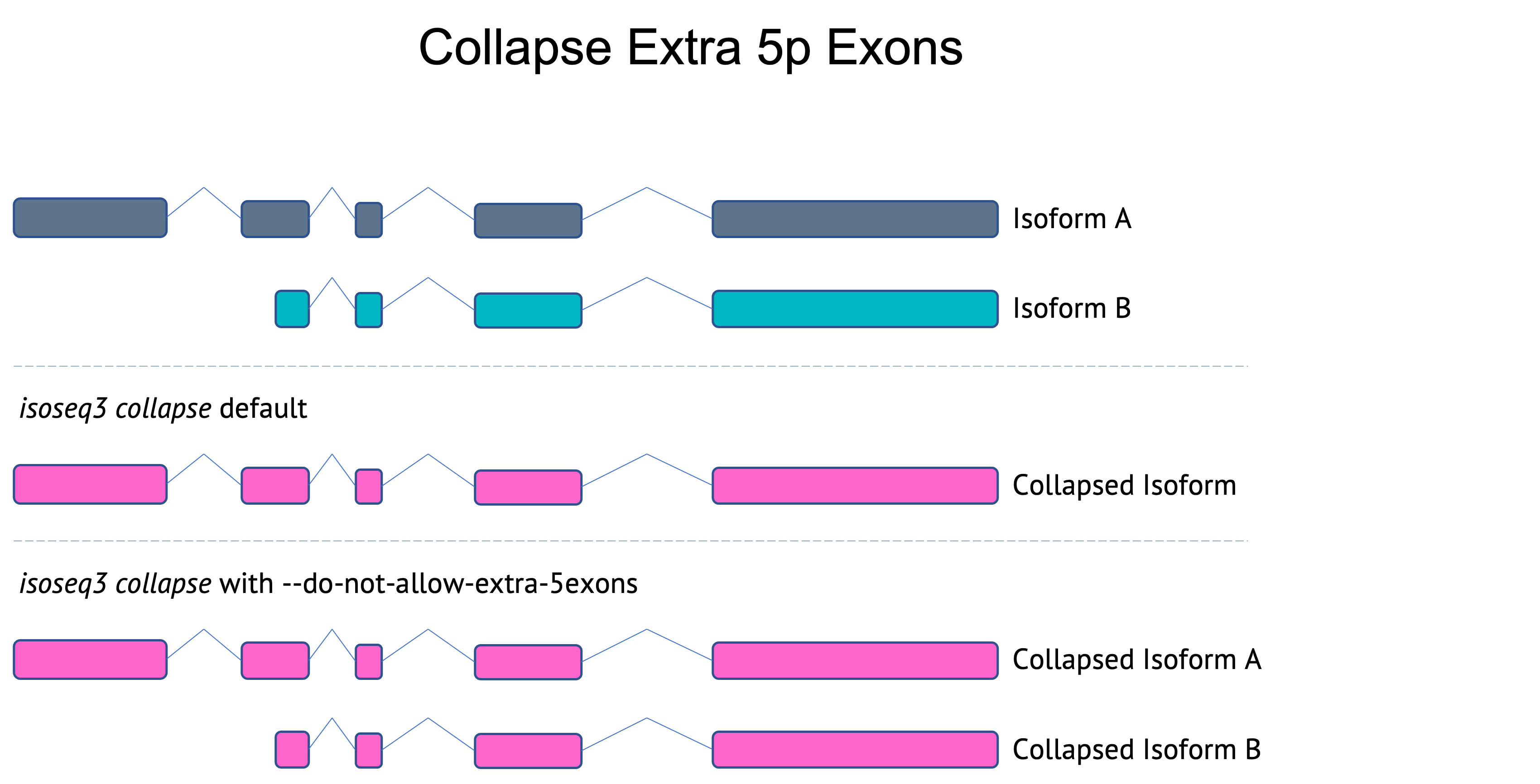
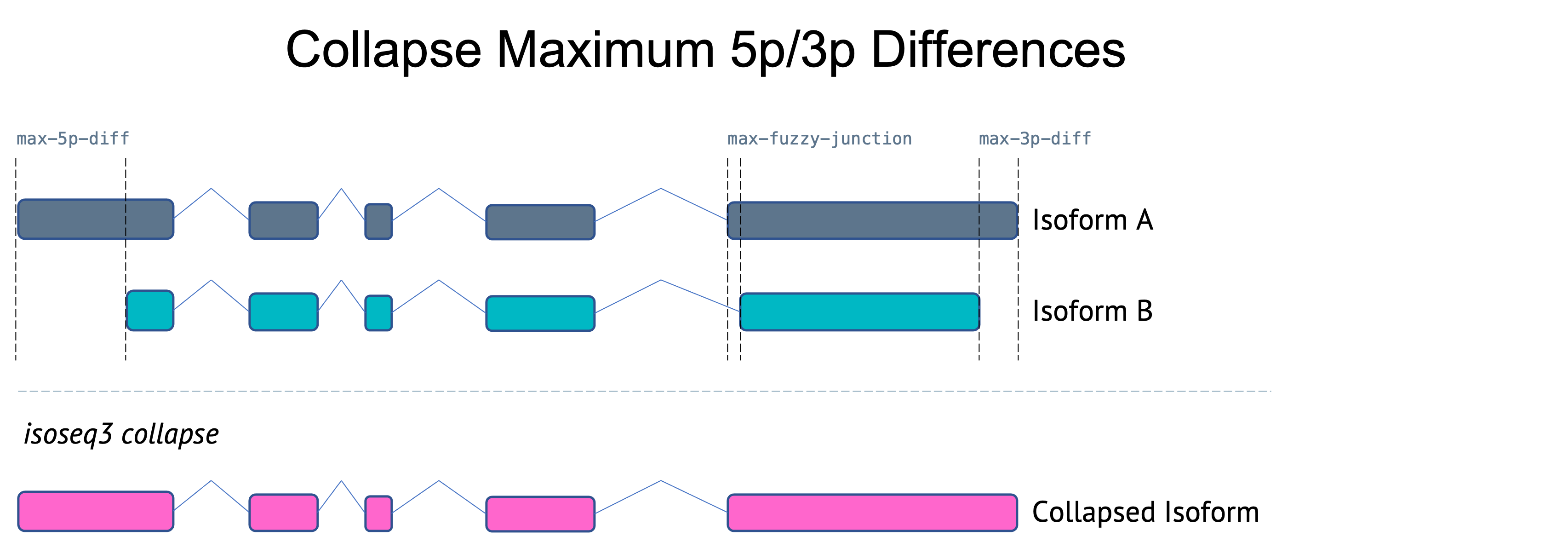
Flexible first/last exon differences
Previous versions of collapse used stringent maximum differences (5bp) for both internal junctions and external junctions. As of v3.8.0, the maximum 5p and 3p differences have been increased and paramaters added to allow adjustments. Note: the maximum 5p difference only applies when --do-not-collapse-extra-5exons is set.
Latest v4.0.0 collapse maximum junction difference parameters:
--max-fuzzy-junction INT Ignore mismatches or indels shorter than or equal to N. [5]
--max-5p-diff INT Maximum allowed 5' difference if on same exon. [50]
--max-3p-diff INT Maximum allowed 3' difference if on same exon. [100]
What if I want to use the legacy collapse logic?
The legacy collapse logic can be recreated using the following parameters:
isoseq collapse --do-not-collapse-extra-5exons --max-5p-diff 5 --max-3p-diff 5 <mapped.bam> <collapsed.gff>