FAQ
Where is the workflow starting from unpolished CCS reads?
To simplify, unify, and future proof Iso-Seq, we decided to remove documentation starting from unpolished CCS reads. With the ever-increasing polymerase read lengths and improvements of CCS, going forward, it is recommended to generate polished CCS reads first and thus make final transcript polishing optional.
How does Iso-Seq v3 compare to versions 1 or 2?
The ever-increasing throughput of the Sequel system gave rise to the need for a scalable software solution that can handle millions of CCS reads, while maintaining sensitivity and accuracy. Internal benchmarks have shown that Iso-Seq v3 is orders of magnitude faster than currently employed solutions and SQANTI attributes Iso-Seq v3 a higher number of perfectly annotated isoforms:
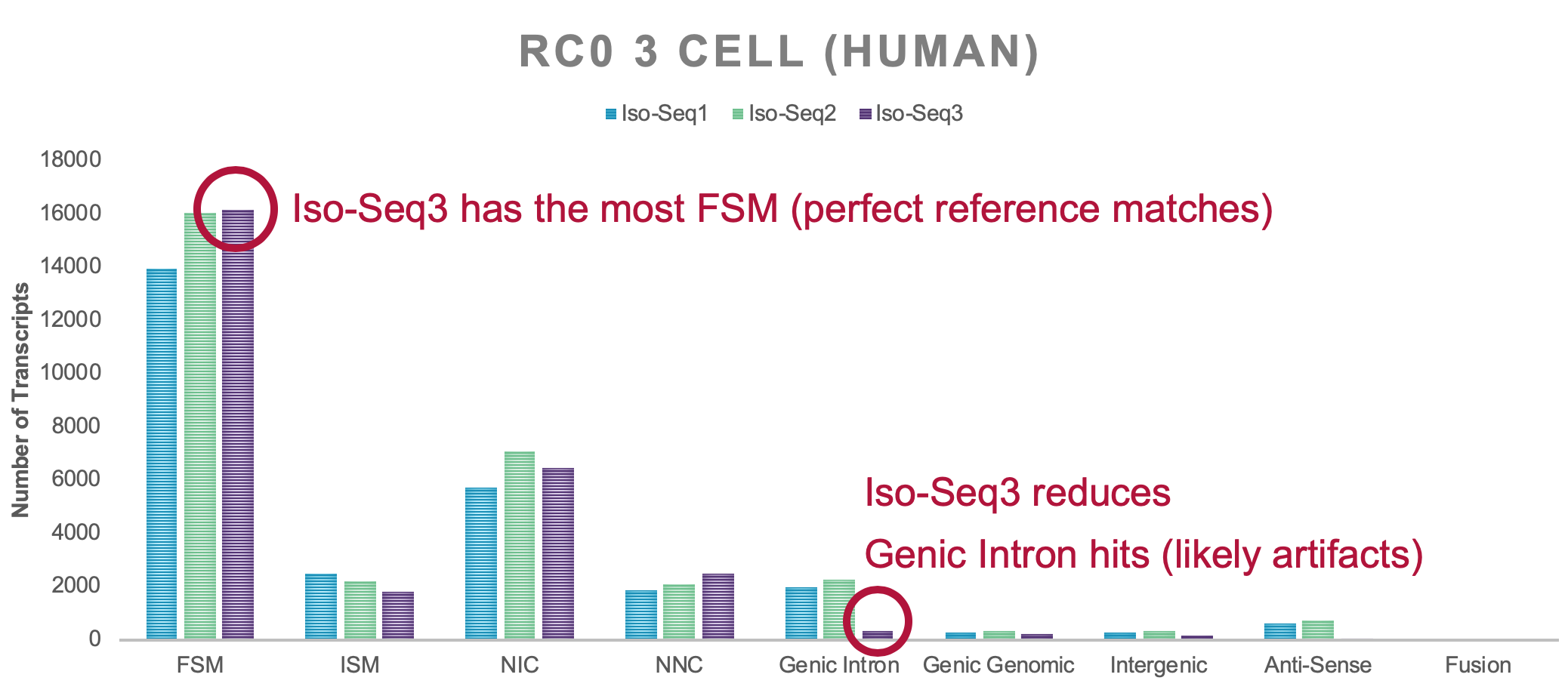
Additional benefit, single linux binary that requires no dependencies.
Why is the number of transcripts much lower with Iso-Seq v3?
Even though we also observe fewer polished transcripts with Iso-Seq v3, the overall quality is much higher. Most of the low-quality transcripts are lost in the demultiplexing step. Iso-Seq v1/2 classify is too relaxed and is not filtering junk molecules to a satisfactory level. In fact, lima calls are spot on and effectively removes most molecules that are wrongly tagged, as in two 5’ or two 3’ primers. Only a proper 5’ and 3’ primer pair allows to identify a full-length transcript and its orientation.
I can’t find the classify step
Starting with version 3.1, classify functionality has been split into two tools. Removal of (barcoded) primers is performed with PacBio’s standard demultiplexing tool lima. Lima does not remove poly(A) tails, nor detects concatemers. For this, isoseq refine generates FLNC reads.
For version 3.0, poly(A) tail removal and concatemer detection is performed in isoseq cluster
My sample has poly(A) tails, how can I remove them?
Use --require-polya for isoseq refine. This filters for FL reads that have a poly(A) tail with at least 20 base pairs and removes identified tail.
How long will it take until my data has been processed?
There is no ETA feature. Depending on the sample type, whole transcriptome or targeted amplification, run time varies. The same number of reads from a whole transcriptome sample can finish clustering in minutes, whereas a single gene amplification of 10kb transcripts can take a couple of hours.
Which clustering algorithm is used?
In contrast to its predecessors, Iso-Seq v3 does not rely on NP-hard clique finding, but uses a hierarchical alignment strategy with O(N*log(N)). Recent advances in rapid alignment of long reads make this this approach feasible.
How many CCS reads are used for the unpolished cluster sequence representation?
Cluster uses up to 10 CCS reads to generate the unpolished cluster consensus.
When are two reads clustered?
Iso-Seq v3 deems two reads to stem from the same transcript, if they meet following criteria:
![]()
There is no upper limit on the number of gaps.