Tertiary analysis of MAS-Seq single cell data with popular community tools
This is a simple tutorial on how to analyze PacBio MAS-Seq single cell data using SMRT Link v11.1. After sequencing, SMRT Link 11.1 has a push button solution to deconcatenate HiFi reads into individual full-length S-reads (transcripts), classify the novelty of isoforms against Gencode and produce a matrix that is compatible with many popular community tools such as Seurat or Scanpy.
SMRT Link 11.1 is available on PacBio’s website. If you prefer to analyze the data with command line, the workflow is described here on https://skera.how/ for read segmentation and https://isoseq.how for single-cell Iso-Seq analysis. We recommend using SMRT Link GUI if possible as it produces many useful plots for sanity check purpose. Nonetheless, the final results from command line analysis are the same as those from SMRT Link.
For this tutorial, we will go through the MAS-Seq single cell demo dataset available here. This is a single-cell PBMC sample containing approximately 5000 input cells. For this analysis, please download the HiFi reads BAM file m64476e_220618_014917.hifi_reads.bam in the folder DATA-SQ2-PBMC_5kcells/0-CCS/.
There are many single-cell analysis packages in the community, and this tutorial does not represent the best ways to analyze MAS-Seq single cell data. However, this shows that you may not need to make a major change in your current bioinformatics workflow for single-cell RNA-seq analysis for many of the routine analysis. You will only have to worry about how to devise ways to discover the new biology in full-length isoforms.
Step-by-step in SMRT Link 11.1
- If this is your first time running MAS-Seq single cell analysis, you’ll first need to download the references required. This can be done by simply clicking on the gear icon on the top right, selecting “About SMRT Link”, followed by “Genomes and Annotations”. By default, SMRT Link supports human and mouse annotations.
- Create a XML file to import the HiFi reads downloaded into SMRT Link (assuming
${SMRT_ROOT}is the installation directory of SMRT Link on your server):${SMRT_ROOT}/smrtcmds/bin/dataset create \ --type ConsensusReadSet \ --name PBMC_5k \ m64476e_220618_014917.consensusreadset.xml \ m64476e_220618_014917.hifi_reads.bam - Go to “Data Management” in SMRT Link, click “Import Data”, select “HiFi Reads (XML)” and select the XML file generated in Step 2. This will import the HiFi reads into SMRT Link.
- You can now go to “SMRT Analysis”, select “Create New Analysis Job”, tick the checkbox on the dataset that has just been imported in Step 3 (It should be the first dataset on top right after the import), then select “Read Segmentations and Single-Cell Iso-Seq” after clicking “Next”.
-
Choose “Human Genome hg38, with Gencode v39” as the “Reference Set”, then start the analysis.
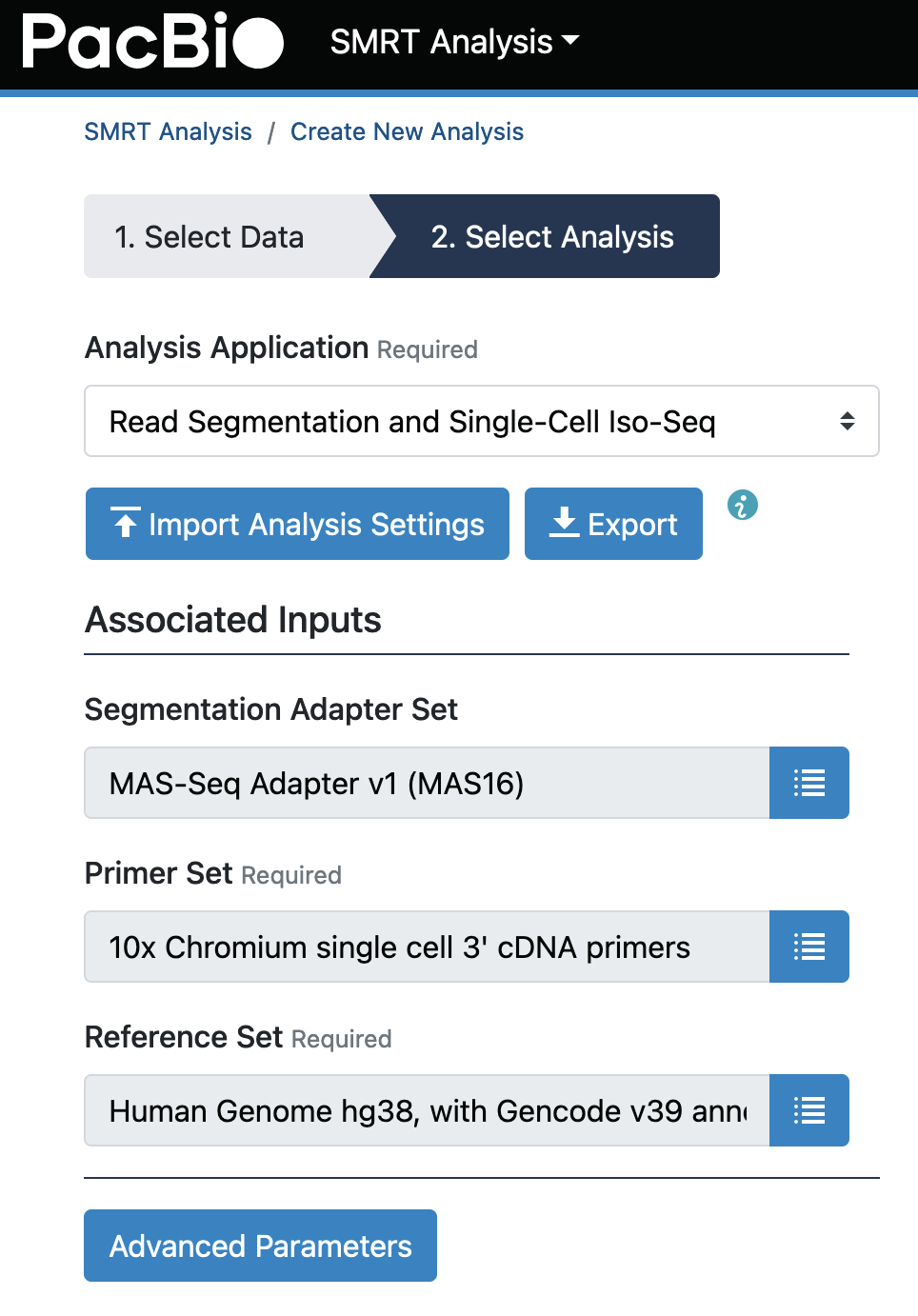
-
After the analysis has finished, you can review the important metrics in SMRT Link GUI. We expect a good MAS-Seq single cell experiment to produce more than 30m S-reads.

- Download the “Single-cell isoform and gene matrix, tar-gzipped” zip file from “File Downloads” tab. Unzip the file to obtain the genes and isoforms matrix in MTX format.
After unzipping the zip file, there are two main folders. genes_seurat contains genes by cells matrix, whereas isoforms_seurat contains isoforms by cells matrix. The matrix format is explained on this 10x Genomics webpage.
Ultrafast cells clustering and labelling with kana
kana is a free web browser tool that allows anyone to analyze single-cell RNA-seq data extremely quickly. As the tool relies on genes level information, we will use the genes by cells matrix (from genes_seurat folder) and pre-process the genes file to make it compatible with kana. Firstly, after downloading the data, get the genes name from the second column of the genes.tsv file. E.g. on command line:
cut -f2 genes.tsv > genes_names.tsv
Go to kana on your web browser, select “Start Analysis”, then use the “Matrix Market” format. Select matrix.mtx for the first input, then genes_names.tsv (second column of genes.tsv, example command above), and finally barcodes.tsv for the barcodes input.
kana should finish within a few minutes, and you will be able to see the TSNE/UMAP plot. If you select cell type annotation in the analysis parameters, you will also be able to see what cells are in your sample. Here’s a screenshot of kana results on the demo dataset, showing the TSNE plot and cell types labelling:
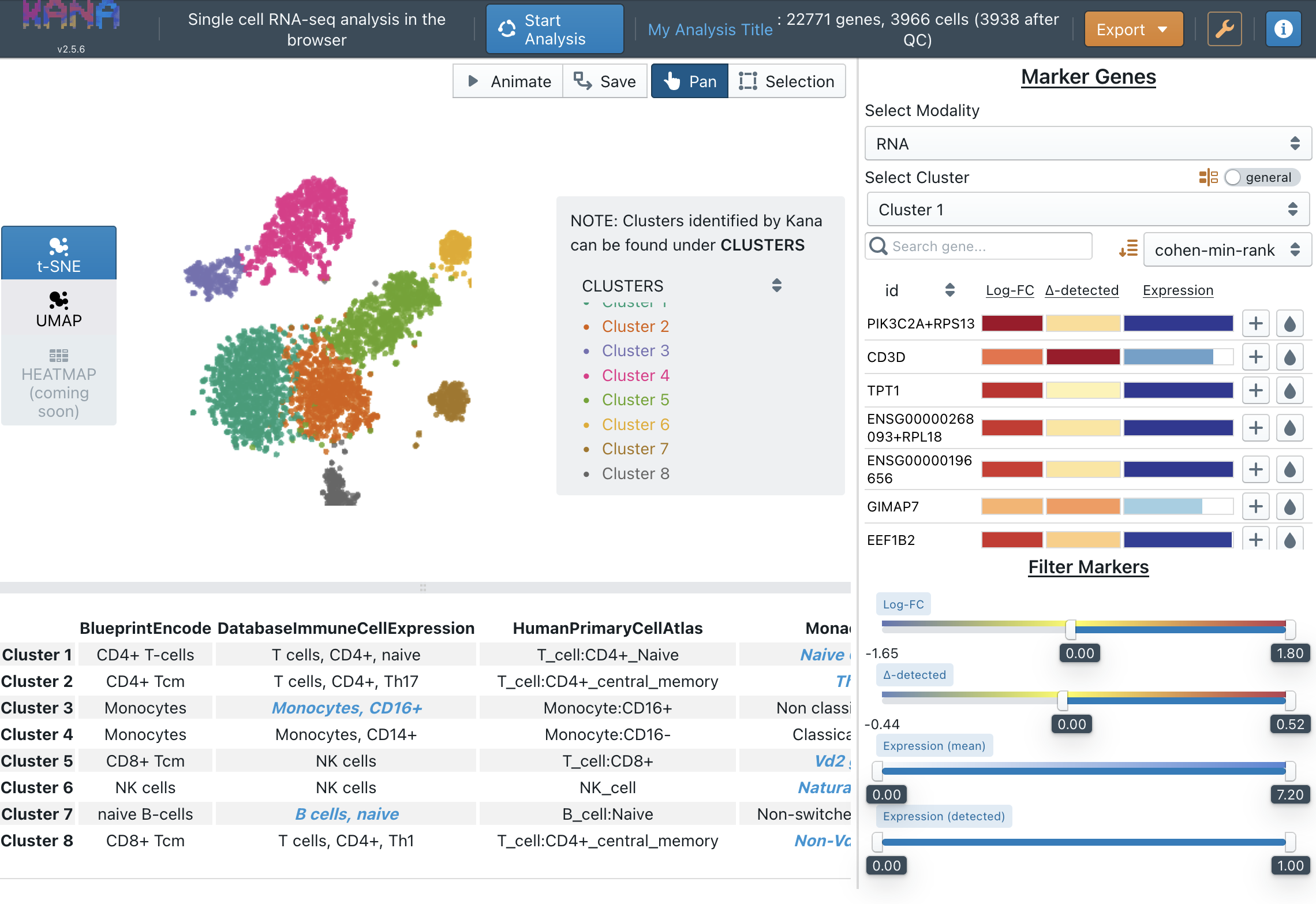
Classifying cell types with CellTypist
While kana is fast and easy, another simple-to-use software for cell types classification is celltypist. This is a Python software that can classify thousands of cells quickly. In addition, it provides many different prediction models meant for different samples.
Similar to many single-cell analysis tools currently available, celltypist works with genes rather than on the isoforms level. A simple pre-processing of the genes TSV file needs to be done to make the files compatible with celltypist (it expects the first column to be gene names):
cut -f2 genes.tsv > genes_names.tsv
# Create a directory for celltypist result
mkdir celltypist_results
If you have Python, celltypist can be installed simply by typing pip install celltypist on the command line. Then, type python on the command line to start a Python interactive shell.
An few lines of codes in the Python interactive shell will classify the matrix from SMRT Link in just a few minutes on a laptop:
import celltypist
from celltypist import models
models.download_models(model = 'Immune_All_Low.pkl')
predictions = celltypist.annotate("matrix.mtx",
model = 'Immune_All_Low.pkl',
transpose_input = True,
gene_file = 'genes_names.tsv',
cell_file = 'barcodes.tsv',
majority_voting=True)
predictions.to_table("celltypist_results")
In the celltypist_results folder, you will find a file named predicted_labels.csv that contains the cell type prediction for each cell. There are many options and models for celltypist, please refer to celltypist website for more details. The cells annotations result should be highly similar to kana. The results from celltypist v1.3.0 for the PBMC 5k dataset using the command above:
CD16+ NK cells 168
Classical monocytes 721
DC2 38
HSC/MPP 15
Memory B cells 71
NK cells 68
Naive B cells 145
Non-classical monocytes 250
Tcm/Naive cytotoxic T cells 98
Tcm/Naive helper T cells 1504
Tem/Effector helper T cells 356
Tem/Temra cytotoxic T cells 279
Tem/Trm cytotoxic T cells 227
pDC 26
Analyzing MAS-Seq single cell data in Seurat
Seurat is a very popular package for single-cell RNA-seq analysis, and we can similarly use it for MAS-Seq single cell data. The guided tutorial from Seurat for PBMC can be used to analyze the gene matrix from MAS-Seq single cell. Briefly, the matrix can be loaded simply with the ReadMtx function from Seurat and you can follow the tutorial along. Note that SMRT Link has already filtered out the ribosomal and mitochondrial genes, so you will not see those genes from the SMRT Link matrix.
An example script based on the tutorial is provided here. The script produces an UMAP plot like this using the PBMC demo data matrix:
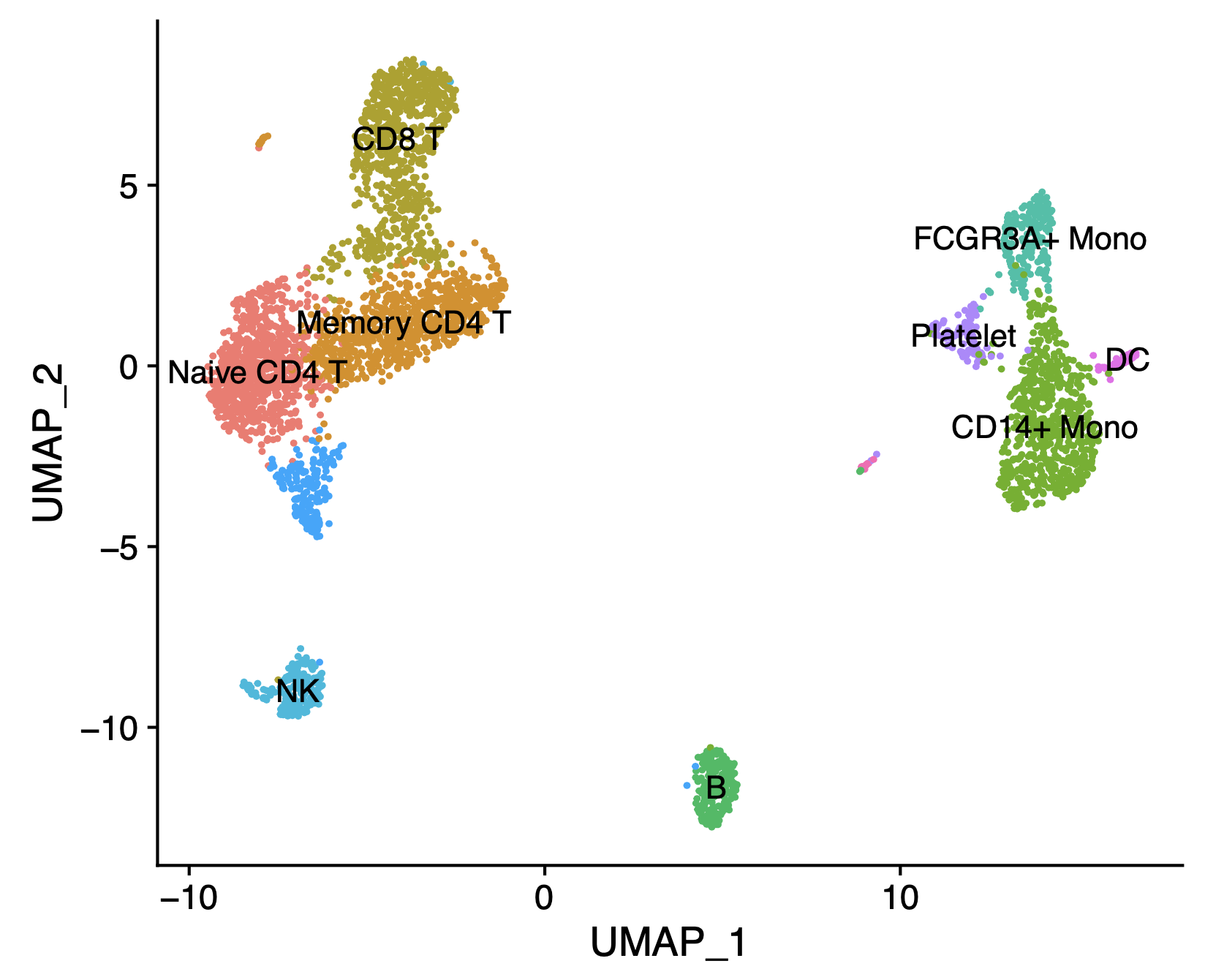
The cell clusters are labelled manually using marker genes following the Seurat tutorial. As you can see, the common PBMC cells are separated well with HiFi sequencing without the need of short-reads sequencing. You can also load the isoforms matrix instead of the genes matrix to discover isoforms unique to any cell type!