Dedup FAQ
NOTE: isoseq groupdedup is now the recommended deduplication tool that replaces the older, slower isoseq dedup. However some documentation figures might still refer to the old isoseq dedup tool as reference.
This FAQ explains how isoseq groupdedup identifies two reads to be from the same founder molecule.
Adjusting maximum mismatches and shifts
The following parameters control the thresholds for mismatches and shifts:
--max-tag-mismatches INT Maximum number of mismatches between tags. [1]
--max-tag-shift INT Tags may be shifted by at maximum of N bases. [1]
In case of unusually short designs (such as a custom UMI that is only 6bp and no cell barcodes), default parameters might lead to overclustering. In this case, please adjust parameters accordingly.
The following is an example of one founder molecule that is sequenced twice. PCR and sequencing errors are introduced, leading to a clipped base in one of the cell barcodes and a substitution in the other cell barcode.
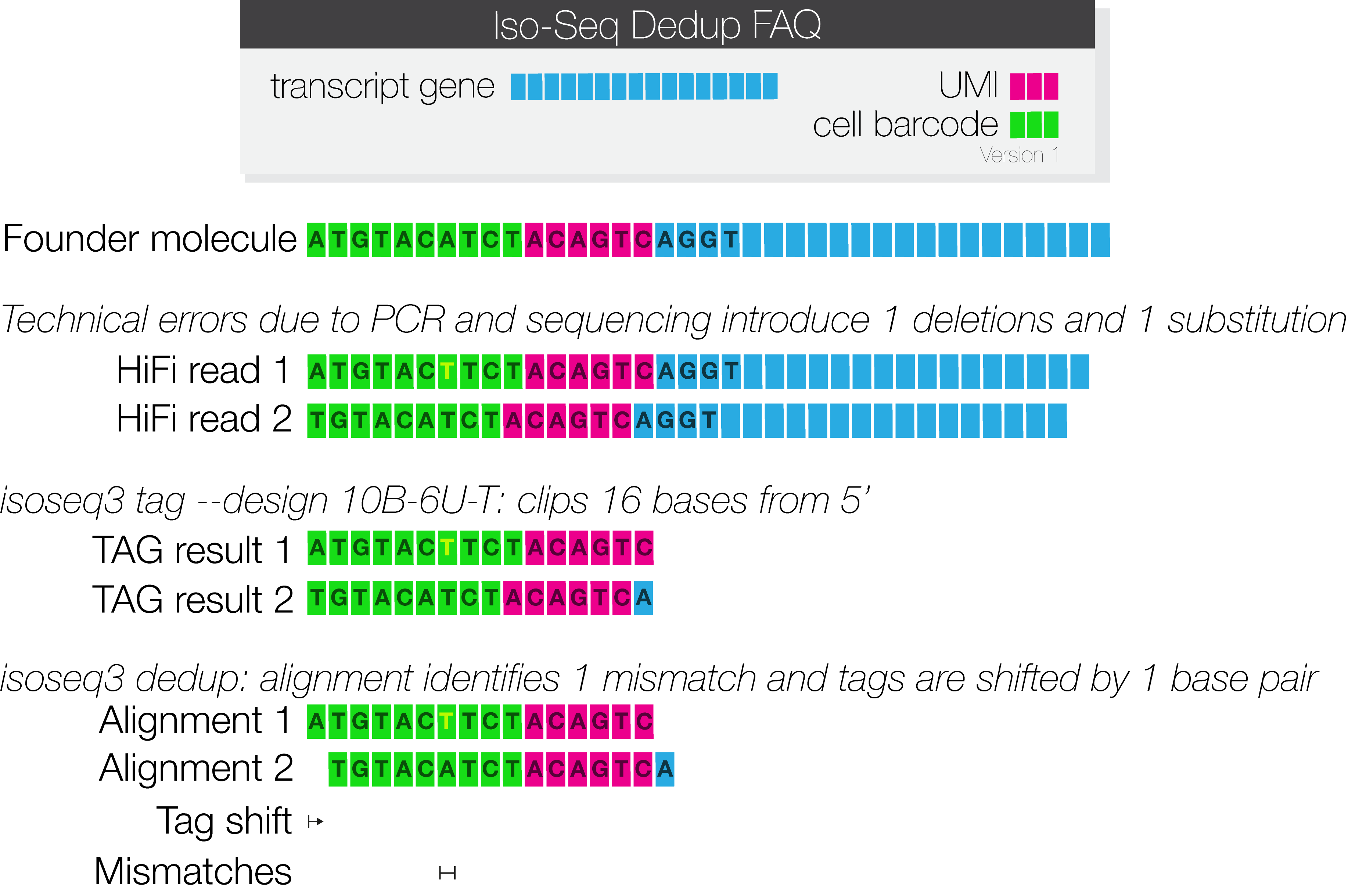
Method
Perform all vs all comparison and cluster two reads if:
- lengths are within +- 50 bp length
- UMI (+cell barcode) match with at max 1 mismatch and may be shifted by at max 1 base
- pairwise concordance is at least 97%
- alignment starts/ends within 5 bp of the other read
- no more than 5 bps are deleted or inserted in a window of 20 bp (like in isoseq cluster)
- groupdedup only: these reads have the same cell barcode
Adjusting insert transcript concordance
The following parameters control the thresholds for how well the inserts (in this case, transcripts) match, even when the UMIs and BCs already match:
--min-concordance-perc INT Minimum insert alignment concordance in %. [97]
--max-insert-gaps INT Maximum number of insert gaps per 20 bp window. [5]
While rare, it is possible to have different transcript molecules share the same UMIs and BCs.
groupdedup only: cell barcode and real cells
If using isoseq groupdedup (which is recommended over isoseq dedup), it can use the corrected cell barcodes from the isoseq correct step for grouping reads.
However, the BAM file must first be sorted by CB tag:
samtools sort –t CB corrected.bam –o corrected.sorted.bam
isoseq groupdedup corrected.bam dedup.bam
Additionally, isoseq groupdedup can use the rc tag from the isoseq correct step and apply to only real cells. This can be turned off with the option below (advanced, not recommended by default):
--keep-non-real-cells Do not skip reads with non-real cells.